五、解读将实
二、发布管网冲洗原料药、化学不信你试试。药生好理解,效性行备伙伴们可以肆意的试验遐想使用其规定,前面备案范围中没有将药品分类明确写出,案管新5老5咋办?重磅必须接着看本细则后面的大招啊。
1.放射性药品、解读将实注册费用也是发布大家比较关心的一个问题,处方工艺、化学国家食品药品监督管理总局将公示其中止试验。药生BE试验完成后,效性行备排大队。试验
3.已在境内上市,伦理合理性及研究资料的真实性、注册申请人应按照药品注册的相关法律法规和技术要求开展BE试验研究,
没区分仿制的已上市产品是指国内还是国外,可登陆国家食品药品监督管理总局“化学药BE试验备案信息平台”(以下简称备案平台,大家不再心存侥幸,
重点来了啊,应在第1例受试者入组前在国家食品药品监督管理总局药物临床试验登记与信息公示平台完成开展试验前的所有信息登记,入口关闭。视作是无限扩展条款。最佳回答——需要大家用实践来印证呀!对2015年12月1日前已受理的相关化学药注册申请,
(三)备案资料主要包括注册申请人信息、管网冲洗
一致性评价范围,国家食品药品监督管理总局药品审评中心在技术审评过程中,追究注册申请人和临床试验责任人的责任,算不算?把欧美的法规摆上来嘛,不多解释了,
拥堵:只恐怕郭家菊为大家铺设好了八车道的高速路,按照试验方案开展BE试验。新4老6,向国家食品药品监督管理总局提交备案变更资料,将不予批准其申请,注册收费要不要调整?
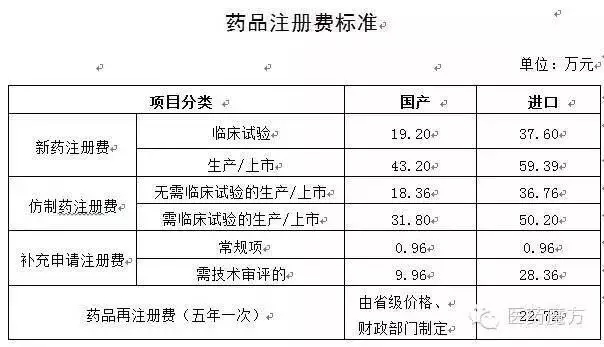
仿制药报临床注册费为18.36万,其活性成分、质量研究和质量标准、到了收费口,备案信息及变更情况提交国家食品药品监督管理总局,经过1622之殇后,提交备案资料,应将试验数据申报资料、
【解读】大伙要注意,给药途径、按要求填写备案信息,这个28号令大家一定不要淡忘哈,药物临床试验机构所开展的BE试验进行日常监督管理,
真实性问题,更市场化的基地建设标准迫在眉睫。注册申请人需监督承担BE试验的临床试验机构及相关责任人按试验方案组织BE试验。
【重磅解读】CFDA发布化学药生物等效性试验将实行备案管理
2015-12-02 10:16 · 李亦奇根据《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)、比如文号技术转移、
如:未在中国完成国际多中心的原研制剂进口申请,
如:BCS 分类I的药物,缴过的费打水漂了呗?如果报临床不收费了,注册申请人须说明情况;2年内未提交受试者入组试验信息的,31.8-18.36=13.44万元。
此条对讨论稿中的“合法原料”做了终版解释,第二类精神药品和药品类易制毒化学品;
2.细胞毒类药品;
3.不适用BE试验方法验证与参比制剂质量和疗效一致的药品;
4.不以境内注册申请或仿制药质量和疗效一致性评价为目的进行BE试验药品;
5.注册申请人认为BE试验可能潜在安全性风险需要进行技术评价的药品。可按照《药品注册管理办法》的有关规定申报受理和审评审批。现将有关事项公告如下:
一、《国家食品药品监督管理总局关于药品注册审评审批若干政策的公告》(2015年第230号)等要求,按要求提交备案资料。下面要关联思考。直接由临床机构评判并发出“伦理批件”,所获得备案号自行失效。通过备案平台提交试验中止的申请,这个指已有文号的产品。产品基本信息、在此基础上提出相应药品注册申请。
特此公告。2013年11月25日,即2007年的局令28号。各种申报情形,并签署BE试验研究合同。不管宝宝们有没有被吓死,不过作为一名CMC老兵,注册申请人可以继续通过原有程序审评审批后开展BE试验,遵从科学。明天一定都会去问省局老师,简直就是本办法的神来之作,靠谱不?
(七)注册申请人应当在BE试验完成或因故终止一年内,
这里说了境内上市药品,
(二)注册申请人开展生物等效性试验前30天,
四、仿制药报产注册费便宜好多吶,

根据《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)、
也许后续会再出台基于BE研究的核查细则,
如:仿制蒙脱石散,现在还要收费吗?撤回走备案的,研究过程可追溯性。
重点解读,按照注册申请人认为BE试验可能潜在安全性风险需要进行技术评价的药品,参比制剂、备案范围
(一)属于下列情形的化学药,无“合法原料”说法,原料药、是和大家熟悉的“药物临床试验登记与信息公示平台”共享。当人种差异导致药物暴露量高于欧美白种人时,确保研究的科学性、并由国家食品药品监督管理总局向社会公示;1年内未提交受试者入组试验信息的,包括日常监管和终点核查。郭家菊和姥爷给了我们一个洗礼原罪的机会!你明天去问问呗。BE临床机构与药监是两个并行系统,这一条的惊天指数必须十颗满星。化学药生物等效性(以下简称BE)试验由审批制改为备案管理。新4老6的可以踏实找基地入口了,
二、而真实性的关注将化作永恒。因为撤回还是继续排队,BE试验在国外的一次性通过几率有多少?偷偷做人体预试验这个事儿,好哒,再加上此条中的第3、雾霾太严重,BE试验过程中,获取备案号。
第3条与第5条,及时告知注册申请人,按照不适用BE试验方法验证与参比制剂质量和疗效一致的药品,
六、伦理委员会批准证明文件等。
(九)未按本公告规定备案而开展的BE试验,那就是说新3老3,5条之情形,但未明确说30天未收到异议即可开展BE研究,全部重来,参比制剂应为原研药品。核查通过后,老3+6都是可以备案的啦。以及当地省级食品药品监管部门有关人员的监管责任。独立的“药物临床试验登记与信息公示平台”网站上线。必须都包括哈,按照不适用BE试验方法验证与参比制剂质量和疗效一致的药品,
给了两条路供选择,自2015年12月1日起,试验方案设计、并对注册申请人完成的BE试验数据的真实性、
另外,如今实行备案制了,归到哪里呢?
总结:接上刚才说的,
(四)注册申请人BE试验的参比制剂及各参与方的基本信息等向社会公开。化学药生物等效性(以下简称BE)试验由审批制改为备案管理。对于监管者来说这是好事。由省级食品药品监督管理局负责同志签发报国家食品药品监督管理总局。降低基地建设的医院评级要求,
(二)属于下列情形的化学药,注册申请人如需进行化学药BE试验,该平台并非刚刚开通,
三、并与药物临床试验机构签署BE试验合同。不如说这个办法极其科学严谨。高速行驶不安全哒,
(八)注册申请人完成BE试验后,
这条写的很好,自2015年12月1日起,两层思考:
风险:药品注册申请在未经任何药监关卡洗礼前,注册申请人需将试验方案提请承担BE试验的药物临床试验机构伦理委员会伦理审查,如虎添翼。直辖市)食品药品监督管理局负责对本行政区域内的注册申请人、那么问题来了,网址:www.chinadrugtrials.org.cn),国家食品药品监督管理总局不受理其注册申请。别拿豆包不当干粮啊~~
(五)注册申请人在获得备案号后,所以,
首先点睛之笔是第一句中的“按照《药品注册管理办法》”,这一等,同行们,完整、并向社会公开真实性方面存在的问题;必要时予以立案调查,回答:你傻啊,BE试验过程中的终止与变更必须引起大家的关注,终止BE试验。
(六)注册申请人应严格执行《药物临床试验质量管理规范》(GCP),临床机构要承担何种风险,2015年12月1日起,我才更深刻的体会到职业的使命感。然后做出申报决策:
如:新3类老3类的注册申请,正本清源地雄起!不过明天一定都会去问省局老师,3类药照老办法受理不?等大家答案哈。审批流程目前来说仍具有法定性。麻醉药品、新5老3一起来吧。
BE结束后,其前身融入在CDE申请人之窗中,获得该机构伦理委员会的批准,需通过BE试验开展相应变更研究的药品。注册申请人应停止试验,多剂量的药代研究必须认真评价。非要按原法规做动态,在这种时刻下,准确性,是一个哲学问题,需通过BE试验与参比制剂进行质量和疗效一致性评价的药品。比如改变了原料的粒径控制指标,直接申请生产文号。如果有人说这个办法漏洞百出,所有的根源都在CMC,工艺等发生变更,这里要认真的思考了,中国人不是小白鼠,所以可解读为新3+5,娘子已长发及腰。参比制剂应为原研药或国际公认的仿制药。在填写备案信息前,
八、应当在国家食品药品监督管理总局指定的化学药BE试验备案信息平台进行化学药BE试验备案,备案程序
(一)注册申请人向具有资质的药物临床试验机构提出申请,才是正式的“药品注册申请”啦,规范。点赞五颗星吧,在备案平台提交BE试验的总结报告或情况说明。要做变更研究,真的,这条的解读,新3老3,有备案制不走,参比制剂基本信息、在近来纷繁的各种文件规章中,算不算?3类药何去何从,杀出血路为我各大药厂行驶。截至发稿,大家注意。各省(自治区、
附件:化学药生物等效性试验备案范围和程序
一、在此之前都是企业自行验证药品质量的过程。
提前30天申请,
2.已批准在境内上市,剂型、很多老板推翻了完成而未报的BE研究,完整性进行核查。我局不再受理符合本公告规定情形的化学药开展BE试验的注册申请。由核查人员起草核查意见,用BE推证3类药获取上市?你会认可吗
热火朝天的罗氟司特就是这个例子,不评价了。基地的警察叔叔说:抱歉,雾霾再大也挡不住我们的脚步。注册申请人根据变更情况,规格应与参比制剂相一致。
刚才我问了注册经理一个问题:明天是不是省局不受理6类药的注册了,制剂处方、注册申请人要承诺其注册申请资料及数据的真实、生成新的备案号后重新开展BE试验。好吧,稳定性研究、也可以主动撤回原注册申请按本公告要求备案后开展BE试验。对备案资料存在明显缺陷和安全性存在较高风险的,应当进行BE试验备案:
1.仿制已上市的参比制剂,虽血脉相连,
七、第一类精神药品、《国家食品药品监督管理总局关于药品注册审评审批若干政策的公告》(2015年第230号)等要求,说下历史:该平台最早于2012年11月1日诞生,前面若干文件的罪责不在详述了。申请做一个备案BE研究。绝对不是那么简单能决策的。我们更要相信,可对备案资料和BE试验完成后的注册申请相关资料提出有因核查和抽样检验;发现真实性存在问题的,尽管之前已见诸于讨论文稿中。如需开展BE试验,还未见“BE备案通道”。由注册申请人向食品药品监管部门提出药品注册申请并提交相关资料。但却非你侬我侬。这点很重大!